C. Campbell, J. R. B. Gomes , M. Fischer, M. Jorge
J. Phys. Chem. Lett., (2018) 9 ,3544–3553.
http://dx.doi.org/10.1021/acs.jpclett.8b00967
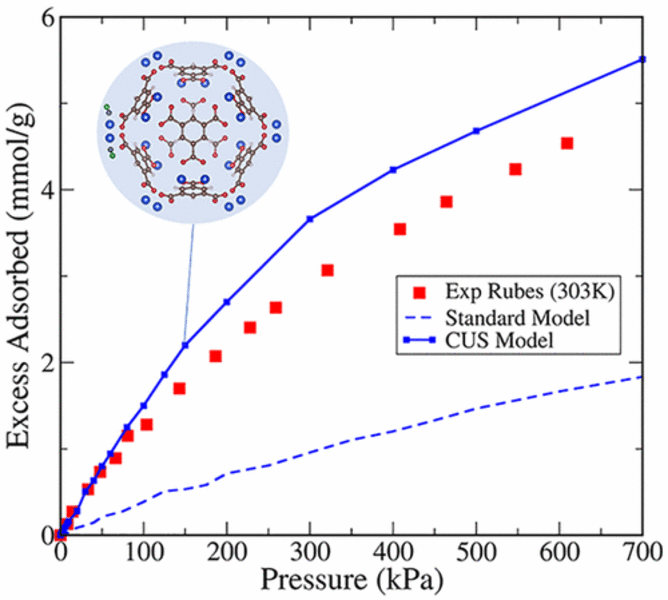
Conventional molecular models fail to correctly describe interactions of adsorbates with coordinatively unsaturated sites (CUS) present in a large number of metal–organic frameworks (MOFs). Here, we confirm the failure of these models for a prototypical polar adsorbate, carbon monoxide, and show that simply adjusting their parameters leads to poor agreement with experimental isotherms when outside the fitting conditions. We propose a new approach that combines quantum mechanical density functional theory (DFT) with Monte Carlo simulations to rigorously account for specific interactions at the CUS. By explicitly including electrostatic interactions and employing accurate DFT functionals that describe dispersion interactions, our modeling approach becomes generally applicable to both polar and nonpolar molecules. We demonstrate that this CUS model leads to substantial improvement in carbon monoxide adsorption isotherm predictions, and correctly captures the coordination binding mechanism. This paper represents a major stepping stone in the development of a robust, transferable and generally applicable approach to describe the complex interactions between gas molecules and CUS, with great potential for use in large-scale screening studies.