S. M. Ayala Mariscal, M. L. Pigazzini, Y. Richter, M. Özel, I. L. Grothaus, J. Protze, K. Ziege, M. Kulke, M. Elbediwi, J.V. Vermaas, L. Colombi Ciacchi, S. Köppen, F. Liu, J. Kirstein.
Nature Communications 13 (2022) , 4692.
https://doi.org/10.1038/s41467-022-32370-5
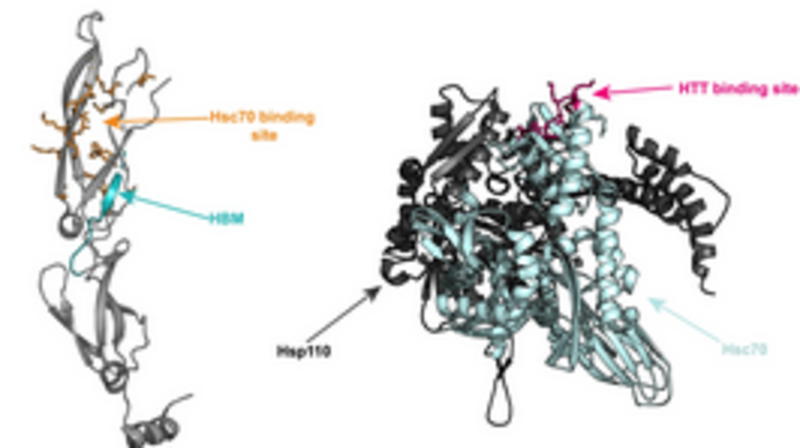
Huntington’s disease is a neurodegenerative disease caused by an expanded polyQ stretch within Huntingtin (HTT) that renders the protein aggregation-prone, ultimately resulting in the formation of amyloid fibrils. A trimeric chaperone complex composed of Hsc70, DNAJB1 and Apg2 can suppress and reverse the aggregation of HTTExon1Q48. DNAJB1 is the rate-limiting chaperone and we have here identified and characterized the binding interface between DNAJB1 and HTTExon1Q48. DNAJB1 exhibits a HTT binding motif (HBM) in the hinge region between C-terminal domains (CTD) I and II and binds to the polyQ-adjacent proline rich domain (PRD) of soluble as well as aggregated HTT. The PRD of HTT represents an additional binding site for chaperones. Mutation of the highly conserved H244 of the HBM of DNAJB1 completely abrogates the suppression and disaggregation of HTT fibrils by the trimeric chaperone complex. Notably, this mutation does not affect the binding and remodeling of any other protein substrate, suggesting that the HBM of DNAJB1 is a specific interaction site for HTT. Overexpression of wt DNAJB1, but not of DNAJB1H244A can prevent the accumulation of HTTExon1Q97 aggregates in HEK293 cells, thus validating the biological significance of the HBM within DNAJB1.
© The Authors licensed under CC BY 4.0